![Room9er ["Room Niner"]:](https://room9er.com/wp-content/uploads/2020/03/cropped-Screen-Shot-2020-03-08-at-3.16.16-PM.png)
How do we slow the rate without lowering the BP? Will slowing the rate even help the BP? How do we raise the BP without speeding up the rate?
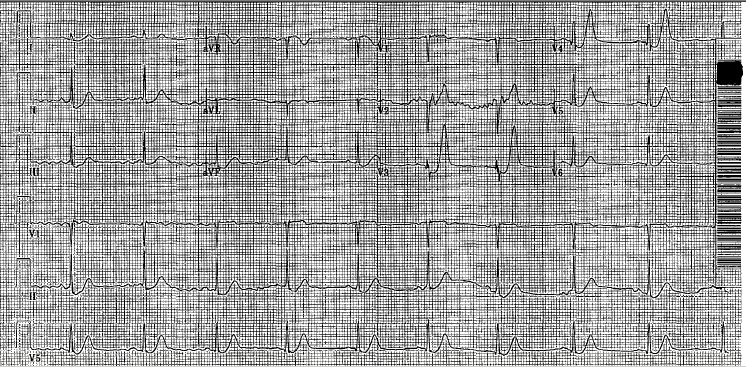
The patient who inspired this post came in for a bowel obstruction. Cards was initially consulted for possible new a-fib, but there really wasn’t much to do from a cardiac standpoint. THEN he perforated his bowel and went for emergency surgery, where he required pressors and went into a-fib with RVR. He was packed open and taken to the SICU, where he was hypotensive to SBP 70s, tachycardic to 160s, and intermittently hypoxic.
If you want to skip my thoughts on the case and head straight for the facts, here is an interesting article about a-fib in critically ill patients. It talks about the various options for management and the pros/cons of each.
If expert opinion is more your style, try this.
Of course, there is also controversy around slowing down a-fib when it is caused by sickness. Should we let the body do its thing to try and compensate? Here’s one article that suggests maybe we shouldn’t get so hung up on rate control in sick people.
Now back to the case. The surgery resident and I were of the mind that slowing the rate and organizing the rhythm should help with cardiac output. There are several reasons why this logic still seems to be in the majority.
– With a-fib you lose atrial kick. That little extra oomph from the atrium may not seem like a big deal, but it can have a significant impact on cardiac output. Here’s a fun article from 1965 that shows a 53% increase in cardiac output from converting a-fib to sinus (using quinidine because 1965). The results have obviously been redemonstrated in more recent studies, but how often do you get to reference quinidine? Not that often.
– Diastolic filling is important for stroke volume. With any tachyarrhythmia, less diastolic filling time means lower stroke volume. However people who do “math” would argue that increasing the rate would likely keep cardiac output about the same. This logic holds up with regular rhythms, but studies show that irregular rhythm decreases cardiac output compared to regular rhythm. Here’s one such study.
– In real life, we went for amiodarone and electrical cardioversion. From the a-fib in critical care article above, it seems like that’s still the best option.
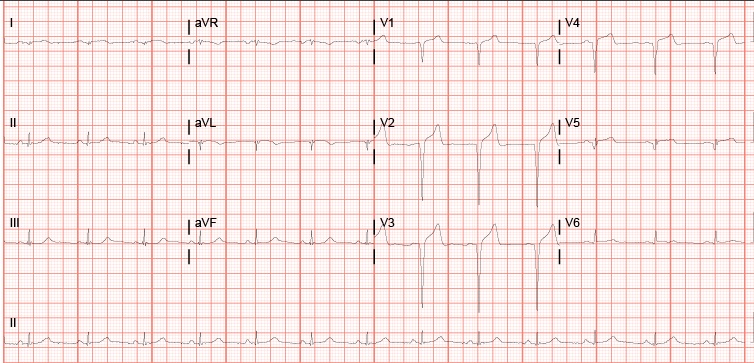
Here’s a good article I wish I had read before embarking on a cluster of a cardioversion. P.S. Put the pads on correctly. Anterior-posterior pads definitely worked better for this 350 pound patient. Anterior pads = fail x3. AP pads = success!
– It’s worth noting that this is not one of the scenarios when you’re worrying about giving someone a stroke with cardioversion. This guy’s risk of death was much greater than his risk of stroke.
Another consideration for this situation was the choice of pressors. Eventually the patient ended up maxed on pretty much all pressors, but that may not always be the case.
Surviving Sepsis guidelines are all about Levophed as a first line pressor, which is usually a great option. But guidelines are just guidelines. How many of our patients are otherwise 100% healthy and just have a little sepsis? Not that many.
– In this case, I think phenylephrine may have been a better first option. Pure alpha agonist activity vasoconstricts without Levophed’s cardiac effects, which probably didn’t do us any favors with the RVR.
– Based on the EMCrit blog above it also seems like phenylephrine might have allowed us to use a beta blocker without worrying about blocking the effects of the pressor or a calcium channel blocker without exacerbating hypotension. Thoughts?
– Anyone have other ideas about pressors in this scenario?
There was a lot more to the case after that, but this post has already ended up way longer than I intended. In the end, it was a 350 pound unhealthy guy with a less than ideal heart, so unfortunately, his family ended up withdrawing care. I doubt anything we could have done would have changed his outcome, but maybe there’s something to learn from it that will help someone else someday. I’ve talked to several people about this case and gotten different opinions from each one, so I figure why not open it up for a few more opinions to really confuse clear things up.